基于SNP进行主成分分析PCA
简介
主成分分析(PCA)是一种线性降维方法,通过线性变换简化数据集,提取关键信息对数据进行区分。群体重测序项目往往能得到百万乃至千万级别的SNP,基于SNP进行PCA的软件有很多,主流是下面三种:
- Plink: https://www.cog-genomics.org/plink/
- GCTA: https://cnsgenomics.com/software/gcta/
- EIGENSOFT: https://github.com/DReichLab/EIG
前面两个软件使用起来相对简单一些,EIGENSOFT运行需要一些配置,相对麻烦一点。
数据准备
我这里使用我以前一篇 文章提到的数据rename.id.maf0.05.geno0.1.vcf,已经进行了过滤。
PCA
这里使用plink以及GCTA进行分析,分析之前都需要数据处理一下:
将vcf数据转换为二进制文件,生成map,ped,bed,bim,fam文件
vcftools --vcf rename.id.maf0.05.geno0.1.vcf --plink --out rename.id.maf0.05.geno0.1.vcf.pca
plink --noweb --file rename.id.maf0.05.geno0.1.vcf.pca --make-bed --out rename.id.maf0.05.geno0.1.vcf.pca_bfile
得到rename.id.maf0.05.geno0.1.vcf.pca_bfile.bed以及rename.id.maf0.05.geno0.1.vcf.pca_bfile.bim
plink计算PCA
这里我们就计算前三个PC,一般也就看前三个PC,当然你可以计算更多,比如前20个
plink --threads 16 --bfile rename.id.maf0.05.geno0.1.vcf.pca_bfile --pca 3 --out rename.id.maf0.05.geno0.1.vcf.pca_bfile
这一步会产生两个文件,一个是以.eigenval结尾的文件,记录特征值,用来计算每个PC所占的比重。另一个是以.eigenvec结尾的文件,记录特征向量
GCTA计算PCA
第一步是计算近交系数,生成grm矩阵
gcta64 --bfile rename.id.maf0.05.geno0.1.vcf.pca_bfile --make-grm --autosome --out rename.id.maf0.05.geno0.1.vcf.pca_bfile_grm
生成rename.id.maf0.05.geno0.1.vcf.pca_bfile_grm.grm.bin,rename.id.maf0.05.geno0.1.vcf.pca_bfile_grm.grm.id以及rename.id.maf0.05.geno0.1.vcf.pca_bfile_grm.grm.N.bin三个文件
第二步计算PCA
gcta64 --grm rename.id.maf0.05.geno0.1.vcf.pca_bfile_grm --pca 3 --out rename.id.maf0.05.geno0.1.vcf.pca_bfile_grm_pca
同样生成以.eigenval和.eigenvec结尾的文件,用于后续绘图。
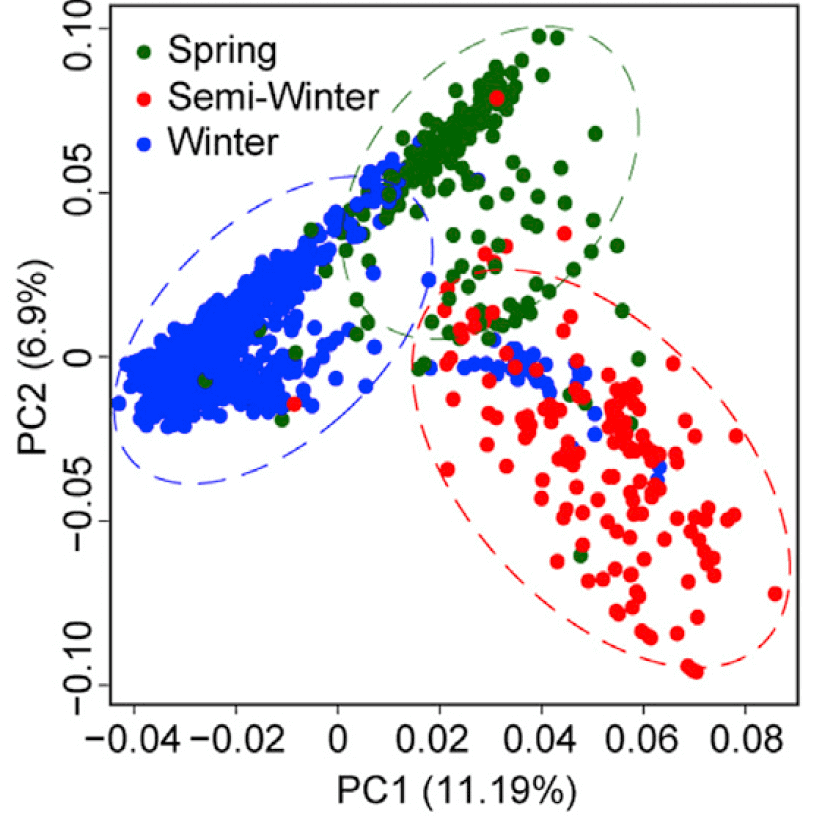
绘图
绘图的话需要再准备样本分类信息sample_info.txt,第一列表示样本名,第二列代表样本的类型
library(tidyverse)
library(export)
pca <- read.table("rename.id.maf0.05.geno0.1.vcf.pca_bfile_grm_pca.eigenvec", header = F)
eigval <- read.table("rename.id.maf0.05.geno0.1.vcf.pca_bfile_grm_pca.eigenval", header = F)
pcs <- paste0("PC", 1:nrow(eigval))
eigval[nrow(eigval),1] <- 0
percentage <- eigval$V1/sum(eigval$V1)*100
eigval_df <- as.data.frame(cbind(pcs, eigval[,1], percentage), stringsAsFactors = F)
names(eigval_df) <- c("PCs", "variance", "proportion")
eigval_df$variance <- as.numeric(eigval_df$variance)
eigval_df$proportion <- as.numeric(eigval_df$proportion)
pc1_proportion <- paste0(round(eigval_df[1,3],2),"%")
pc2_proportion <- paste0(round(eigval_df[2,3],2),"%")
sample <- read.table("sample_info.txt", header = F)
data <- left_join(pca,sample,by="V1")
data <- data[,-2]
colnames(data) <- c("Sample","PC1","PC2","PC3","Type")
data$Type <- factor(data$Type, levels = c("Group1","Group2","Group3"))
p_pca <- ggplot(data,aes(PC1,PC2))+
geom_point(aes(color=Type), size=3)+
stat_ellipse(aes(color=Type),level = 0.95, show.legend = FALSE, size=1)+
scale_color_manual(values = c("#2a6117","#e93122","#0042f4"))+
theme(panel.grid = element_blank(),
panel.background = element_blank(),
panel.border = element_rect(fill = NA, colour = "black"),
legend.title = element_blank(),
legend.key = element_blank(),
axis.text = element_text(colour = "black", size=12, family = "Times New Roman"),
axis.title = element_text(color="black",size = 15, family = "Times New Roman"),
legend.text = element_text(colour = "black", size=15, family = "Times New Roman"),
legend.position = c(0.15,0.15)
)+
labs(x=paste0("PC1(",pc1_proportion,")"),
y=paste0("PC2(",pc2_proportion,")"))
graph2office(file = "pca.pptx")
最后直接导出到PPT中用于编辑。