使用SNPhylo基于SNP构建群体系统发育树
简介
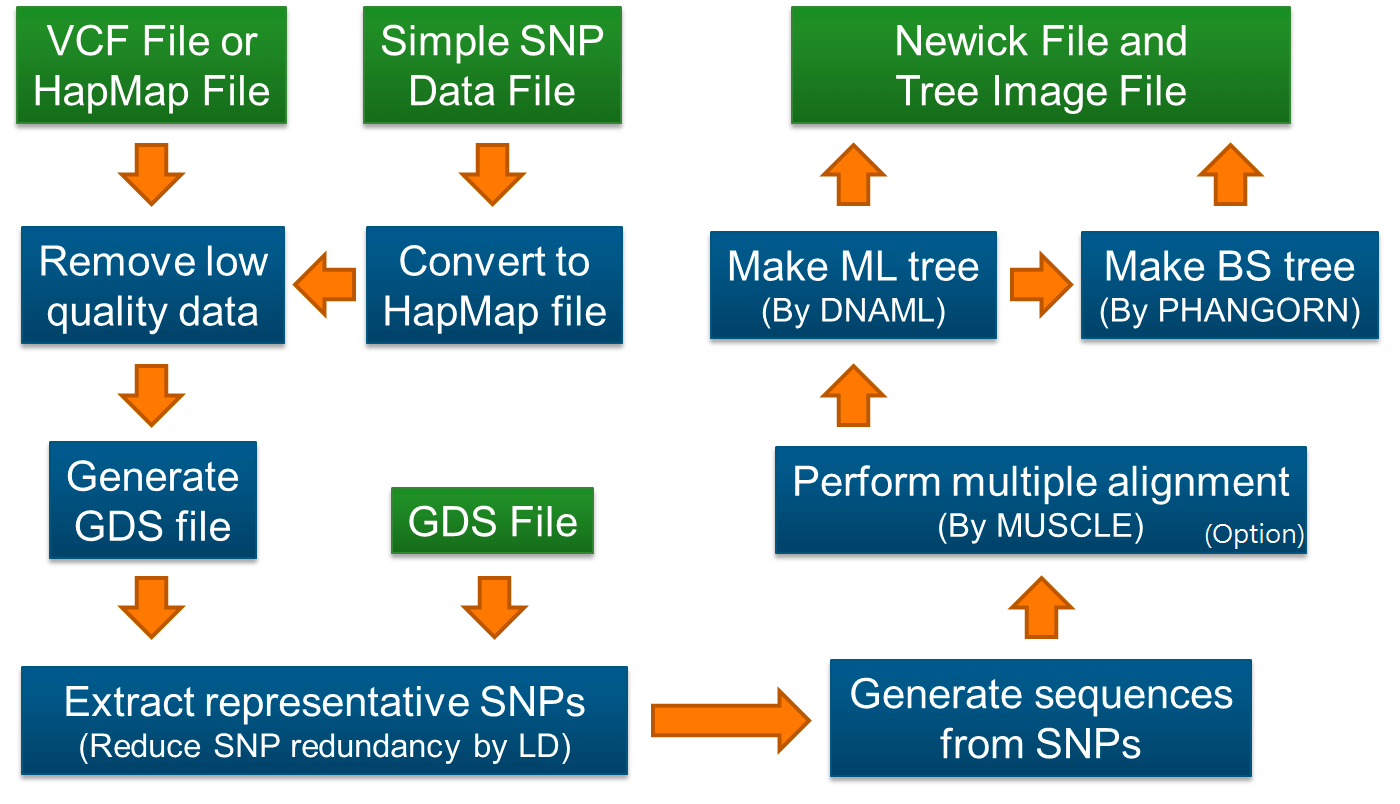
系统发育树是一种推断各种生物之间进化关系的好方法,在进化研究中得到了广泛的应用,得益于测序技术的发展以及成本的不断下降,大量的物种以及群体被测序,产生了海量的基因型数据,在重测序项目中,基于SNP数据进行系统发育树的构建有利于更全面地囊括整个基因组层面的变异进行分析。 SNPhylo是基于SNP数据构建群体系统发育树的一个pipeline,可以接受主流的VCF文件以及HapMap文件,同时其内置了过滤功能,通过对低质量的SNP,连锁不平衡进行过滤,生成用于构建群体系统发育树的输入数据,SNPhylo会调用muscle进行对序列比对,DNAML构树,具体流程如下:
SNPhylo有以下特性:
-
基于全基因组SNP的树状结构。传统的建树方法是基于单拷贝基因,核糖体RNA基因,内部转录间隔序列(ITS)等具有特定属性的基因,而SNPhylo则是从全基因组层面构建进化树,因此更加准确;
-
通过连锁不平衡减少SNP的冗余。同一LD Block中的SNP提供了冗余的系谱信息,SNPhylo在一个LD Block只保留一个有效的SNP,在不丢失信息位点的情况下,大大减少了运行时间;
-
树的构建过程是高度自动化的。SNPhylo可以接受主流的VCF文件作为直接输入数据,一行命令就可以生成最大似然树。
SNPhylo的源代码在https://github.com/thlee/SNPhylo
数据准备
SNPhylo内置了一系列的过滤功能,但是我倾向于自己先过滤数据,这样后续SNPhylo运行时间也快些。
maf以及geno过滤
过滤之前先处理一下基因型数据,很多软件是为人体研究设计的,不支持带有特殊字符的染色体名称,比如chr1,因此第一步我们先修改染色体名称为数值型,这里我们利用bcftools:
# 先利用bgzip压缩,tabix构建索引
bgzip < original.vcf > original.vcf.gz
tabix -p vcf original.vcf.gz
# 制作染色体名称对应文件
echo "chr1 1" >> chr_name_conv.txt
echo "chr2 2" >> chr_name_conv.txt
....
## 染色体名称修改
bcftools annotate --rename-chrs chr_name_conv.txt original.vcf.gz | bgzip > rename.vcf.gz
tabix -p vcf rename.vcf.gz
## 添加SNP ID, GATK出来的VCF文件是没有SNP ID的,这里还是利用bcftools添加ID
bcftools annotate --set-id '%CHROM\_%POS\_%REF\_%FIRST_ALT' rename.vcf.gz |bgzip > rename.id.vcf.gz
tabix -p vcf rename.id.vcf.gz
only include SNPs with MAF >= 0.05 and include only SNPs with a 90% genotyping rate (10% missing) use
plink --vcf rename.id.vcf.gz --maf 0.05 --geno 0.1 --recode vcf-iid --out rename.id.maf0.05.geno0.1
下面根据LD进行过滤并提取符合条件的SNP用于建树
## LD标记
plink --vcf rename.id.maf0.05.geno0.1.vcf --indep-pairwise 50 10 0.2 --out rename.id.maf0.05.geno0.1
## LD过滤提取
plink --extract rename.id.maf0.05.geno0.1.prune.in --make-bed --out rename.id.maf0.05.geno0.1.prune.in --recode vcf-iid --vcf rename.id.maf0.05.geno0.1.vcf
此时我们得到了rename.id.maf0.05.geno0.1.prune.in.vcf用作SNPhylo的输入数据。
构建进化树
SNPhylo需要安装一些依赖:
- R包(getopt, phangorn, gdsfmt,SNPRelate):用于过滤等
- python2.7
- muscle:序列联配,可选
- PHYLIP:构建进化树
安装这些环境之后放入环境变量中,或者一会配置的时候手动输入这些软件的位置
此时从GitHub下载最新的SNPhylo
git clone https://github.com/thlee/SNPhylo.git
配置:
cd SNPhylo
bash setup.sh
# 配置信息
Version: 20141127
START TO SET UP FOR SNPHYLO!!!
The detected path of R is /usr/bin/R. Is it correct? [Y/n] y
The detected path of python is /usr/bin/python. Is it correct? [Y/n] y
The detected path of muscle is /home/taoyan/biosoft/mybin/muscle. Is it correct? [Y/n] y
The detected path of dnaml is /home/taoyan/biosoft/phylip-3.697/exe/dnaml. Is it correct? [Y/n] y
At least one R package (gdsfmt, SNPRelate, getopt or phangorn) to run this pipeline is not found. Are the packages already installed? [y/N] y
Please enter the directory for R packages (ex: /home/foo/r_packages): /database/R_Library
SNPHYLO is successfully installed!!!
看一下使用说明:
$ ./snphylo.sh -h
Determine phylogenetic tree based on SNP data with a VCF, a HapMap, a Simple SNP or a GDS file
Version: 20180901
Usage:
snphylo.sh -v VCF_file [-p Maximum_PLCS (5)] [-c Minimum_depth_of_coverage (5)]|-H HapMap_file [-p Maximum_PNSS (5)]|-s Simple_SNP_file [-p Maximum_PNSS (5)]|-d GDS_file [-l LD_threshold (0.1)] [-m MAF_threshold (0.1)] [-M Missing_rate (0.1)] [-o Outgroup_sample_name] [-P Prefix_of_output_files (snphylo.output)] [-b [-B The_number_of_bootstrap_samples (100)]] [-a The_number_of_the_last_autosome (22)] [-t The_number_of_cores_used (1)] [-r] [-A] [-h]
Options:
-A: Perform multiple alignment by MUSCLE
-b: Perform (non-parametric) bootstrap analysis and generate a tree
-h: Show help and exit
-r: Skip the step removing low quality data (-p and -c option are ignored).
Acronyms:
PLCS: The percent of Low Coverage Sample
PNSS: The percent of Sample which has no SNP information
LD: Linkage Disequilibrium
MAF: Minor Allele Frequency
Simple SNP File Format:
#Chrom Pos SampleID1 SampleID2 SampleID3 ...
1 1000 A A T ...
1 1002 G C G ...
...
2 2000 G C G ...
2 2002 A A T ...
...
很简单,直接将准备好的rename.id.maf0.05.geno0.1.prune.in.vcf输进去记好了(我这里无需过滤了,所以设置了参数-r):
./snphylo.sh -v rename.id.maf0.05.geno0.1.prune.in.vcf -A -b -B 1000 -r
运行结束后回生成一系列文件:
snphylo.output.bs.png
snphylo.output.bs.tree
snphylo.output.fasta
snphylo.output.filtered.vcf
snphylo.output.gds
snphylo.output.id.txt
snphylo.output.ml.png
snphylo.output.ml.tree
snphylo.output.ml.txt
snphylo.output.phylip.txt
snphylo.tar.gz
其中有树文件,图片也有,但是丑,以前有iTOL,现在收费了,还是老老实实用Y叔的ggtree自己绘制美化吧