EDTA:转座子注释
简介
转座子注释目前有非常多的软件,而EDTA(The Extensive de novo TE Annotator)整合了大部分目前常用的转座子注释软件,值得注意的是EDTA是全基因组从头注释。EDTA最大的好处就是简单实用,而且经过大量的改进,目前在安装、运行以及结果解释等方面十分完善。
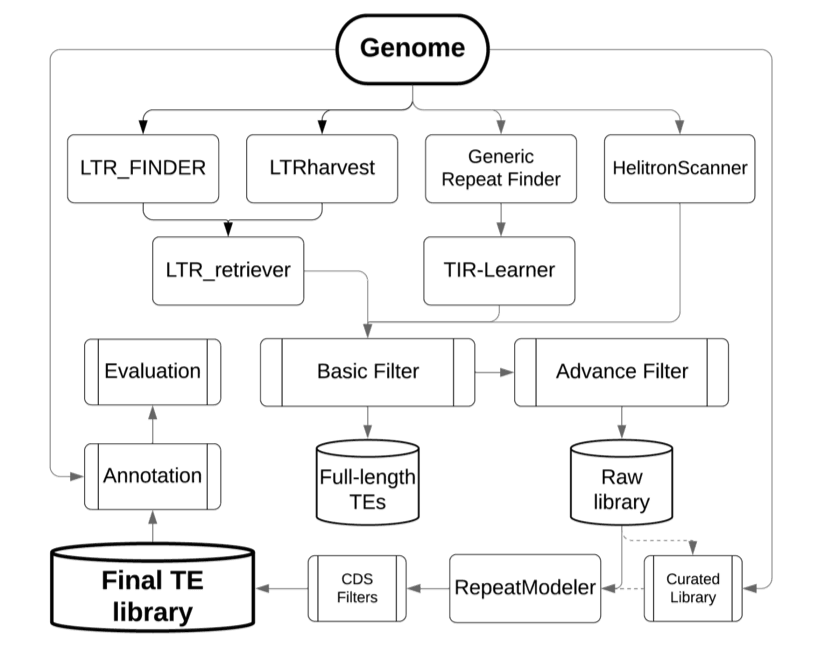
流程
具体可以参考EDTA的 GitHub主页以及发表在Genome Biology上的 文章
安装
EDTA提供了四种安装方式:
Quick installation using conda (Linux64)
conda install -c bioconda -c conda-forge edta
Quick installation using Singularity (good for HPC users)
Installation:
singularity build --sandbox EDTA.sif docker://kapeel/edta
Usage:
singularity exec {path}/EDTA.sif /EDTA/EDTA.pl --genome genome.fa [other parameters]
{path} is the path you build the EDTA singularity image
Quick installation using Docker (good for root/Mac users)
Installation:
docker pull kapeel/edta
Usage:
docker run -v $PWD:/in -w /in kapeel/edta --genome genome.fa [other parameters]
Step by step installation using conda
conda create -n EDTA
conda activate EDTA
conda config --env --add channels anaconda --add channels conda-forge --add channels bioconda
conda install -n EDTA -y cd-hit repeatmodeler muscle mdust blast openjdk perl perl-text-soundex multiprocess regex tensorflow=1.14.0 keras=2.2.4 scikit-learn=0.19.0 biopython pandas glob2 python=3.6 tesorter genericrepeatfinder genometools-genometools ltr_retriever ltr_finder numpy=1.16.4
git clone https://github.com/oushujun/EDTA
./EDTA/EDTA.pl
文件
只需要提供基因组文件就行了,但是需要注意的是序列名称不能超过15个字符,所以简单点好不易出错。如果你研究的物种还有以下文件的话也可以提供,可以提高注释的准确性:
- 所研究物种或者亲缘关系比较近的物种的CDS文件(需要剔除掉内含子以及UTRs)
- 基因坐标信息
- 可信度非常高的TE库,无需全基因组完整的,部分零散的也行,但是需要注意的是可信度必须是比较高的,不然就不要提供了,免得适得其反
用法
用法非常简单,毕竟整合了大部分主流软件且封装到一起
一步到位From head to toe
You got a genome and you want to get a high-quality TE annotation:
perl EDTA.pl [options]
--genome [File] The genome FASTA
--species [Rice|Maize|others] Specify the species for identification of TIR candidates. Default: others
--step [all|filter|final|anno] Specify which steps you want to run EDTA.
all: run the entire pipeline (default)
filter: start from raw TEs to the end.
final: start from filtered TEs to finalizing the run.
anno: perform whole-genome annotation/analysis after TE library construction.
--overwrite [0|1] If previous results are found, decide to overwrite (1, rerun) or not (0, default).
--cds [File] Provide a FASTA file containing the coding sequence (no introns, UTRs, nor TEs) of this genome or its close relative.
--curatedlib [file] Provided a curated library to keep consistant naming and classification for known TEs.
All TEs in this file will be trusted 100%, so please ONLY provide MANUALLY CURATED ones here.
This option is not mandatory. It's totally OK if no file is provided (default).
--sensitive [0|1] Use RepeatModeler to identify remaining TEs (1) or not (0, default).
This step is very slow and MAY help to recover some TEs.
--anno [0|1] Perform (1) or not perform (0, default) whole-genome TE annotation after TE library construction.
--rmout [File] Provide your own homology-based TE annotation instead of using the EDTA library for masking. File is in RepeatMasker .out format. This file will be merged with the structural-based TE annotation. (-anno 1 required). Default: use the EDTA library for annotation.
--evaluate [0|1] Evaluate (1) classification consistency of the TE annotation. (-anno 1 required). Default: 0.
This step is slow and does not affect the annotation result.
--exclude [File] Exclude bed format regions from TE annotation. Default: undef. (-anno 1 required).
--threads|-t [int] Number of theads to run this script (default: 4)
--help|-h Display this help info
参数说明非常详细,用起来十分清爽
分类注释
也可以指定需要注释的转座子类型:
perl EDTA_raw.pl [options]
--genome [File] The genome FASTA
--species [Rice|Maize|others] Specify the species for identification of TIR candidates. Default: others
--type [ltr|tir|helitron|all] Specify which type of raw TE candidates you want to get. Default: all
--overwrite [0|1] If previous results are found, decide to overwrite (1, rerun) or not (0, default).
--threads|-t [int] Number of theads to run this script
--help|-h Display this help info
另外还支持断点运行,中间软件运行意外终止的话,可以从终止那一步继续运行
Finish the rest of the EDTA analysis (specify -overwrite 0 and it will automatically pick up existing results in the work folder)
perl EDTA.pl --overwrite 0 [options]
结果
返回的结果非常多,并且分门别类帮你整理好,具体我就不讲了,毕竟文档讲得太详细了。