Nature Reviews Genetics | 测序时代的基因组转座子
简介
许多物种的基因组大部分是来自于转座子(Transposable elements, TEs)。此外,通过各种自我复制机制,TEs在大多数物种基因组中持续增殖。TEs会影响调控、转录以及新蛋白形成进而与性状比如疾病相关联。尽管TE表现出了显著的影响,但是很多基因组研究还是将它剔除在外,这主要是由于它具有高度的序列重复性,导致研究变得十分复杂。幸运的是随着大量的方法以及计算机软件的开发,TE的研究逐渐深入。本综述总结了TE研究的相关计算工具并着重指出了未来的TE研究存在的挑战与空白。对TE进行基因组上全面的分析不仅仅是“掩盖”这些重复序列(This Review presents a summary of computational resources for TEs and highlights some of the challenges and remaining gaps to perform comprehensive genomic analyses that do not simply “mask” repeats)。
转座子
转座子首先由Barbara McClintock于1948年在玉米中发现,之后在所有动物和植物以及各种真核生物中陆续发现。转座子可以通过不同的机制从基因组的一个位置"跳跃"到另一个位置,这种"跳跃"可以发生在同一条染色体内,也可以发生在不同染色体间。转座是转座子的标志性特征,转座子从供给位点(Donor site)整合到目标位点(Target site)被称为一个完整的转座过程。转座子可以通过两种机制进行转座:
- 复制后粘贴(Copy-and-paste)
- 剪切后粘贴(Cut-and-paste)
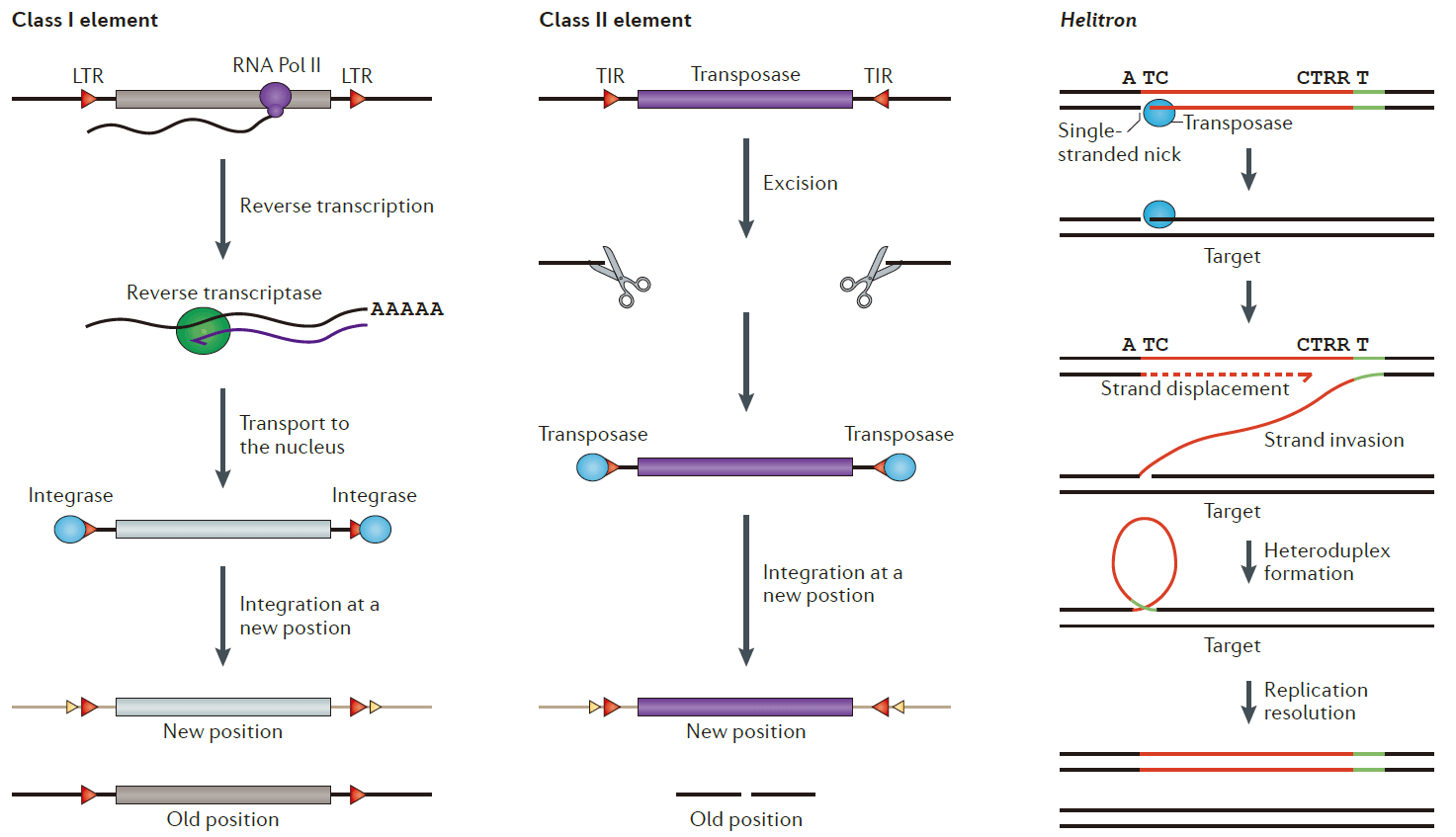
复制后粘贴
首先RNA聚合酶Ⅱ(RNA polymerase Ⅱ)将转座子转录成mRNA,再由逆转录酶逆转录形成一个自身DNA(cDNA)拷贝,最后通过整合酶(Integrase)将拷贝整合到目标位点。这一过程RNA起到了一个媒介的作用,所以这一类转座子称为RNA类转座子,也被称为Ⅰ型转座子或逆转录转座子(Retrotransposon)。
剪切后粘贴
转座子不以RNA为媒介,直接被转座酶(Transposase)从供给位点脱离出来通过整合酶整合到目标位点。有一个特例是“滚环”机制,通过Y2转座酶和整合酶实现链入侵,随后通过链置换(Strand displacement),DNA复制,异源双链形成将转座子整合到目标位点,这些转座子在转座过程中都不需要RNA为媒介,所以这些转座子被称为DNA转座子(DNA transposon)或Ⅱ型转座子。
Helitrons转座子
Helitrons 转座子是近年来发现的一种新型 DNA 转座子(上面提到的“滚环”机制),最初是利用基于重复序列的计算方法在拟南芥基因组中鉴定出来的。后来发现,大多数植物和许多动物基因组中都携带 Helitrons 转座子。Helitrons 转座子具有典型的 5’TC 以及 3’CTRR(R为A或G)末端,并在3’末端上游约 15~20bp 处有一个茎环结构,是转座子的终止信号。Helitrons 转座子转座后,通常插入 AT-rich 区域的 AT 靶位点。和反转座子和转座子不同,Helitrons 通过滚环(rolling circle)的方式进行转座。并且,在滚环复制的转座过程中经常捕获和携带基因片段,可导致基因拷贝数的变化,也会在一定程度上促进基因组的进化。
Maverick转座子
Maverick类型的重复序列一般较大,可达到10-20Kb,两端含有较长的TIR序列,在除植物之外的真和生物中广泛存在。其编码的蛋白可达到11个,但数目和顺序有所不同,有些蛋白与DNA病毒有一定的同源性。这些蛋白中的DNA聚合酶B和整合酶(c-int type)与反转录转座子中的相关蛋白类似,但同样不含有逆转座酶。
这几类转座子具体转座机制如下:
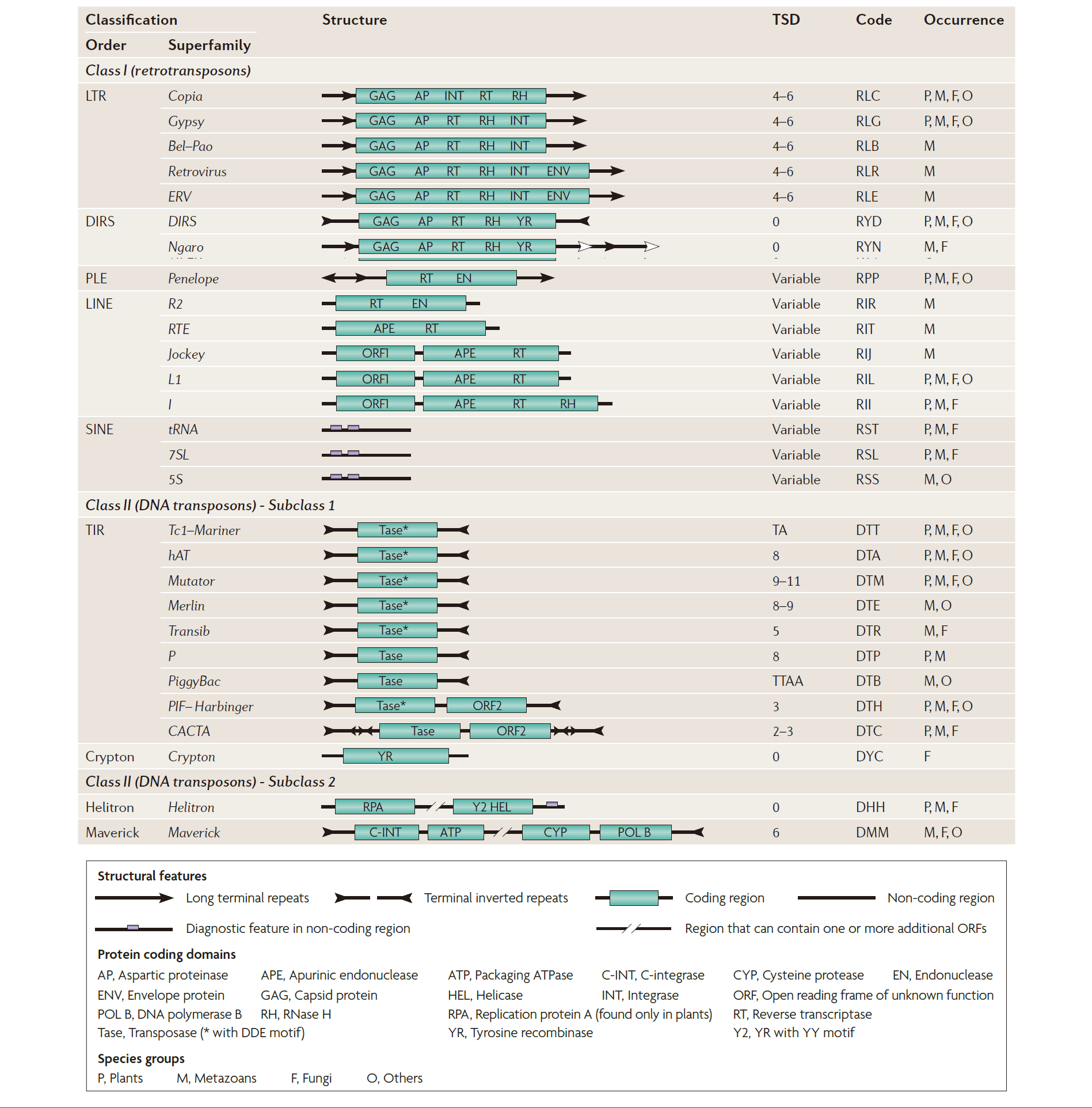
TEs具体分类如下:
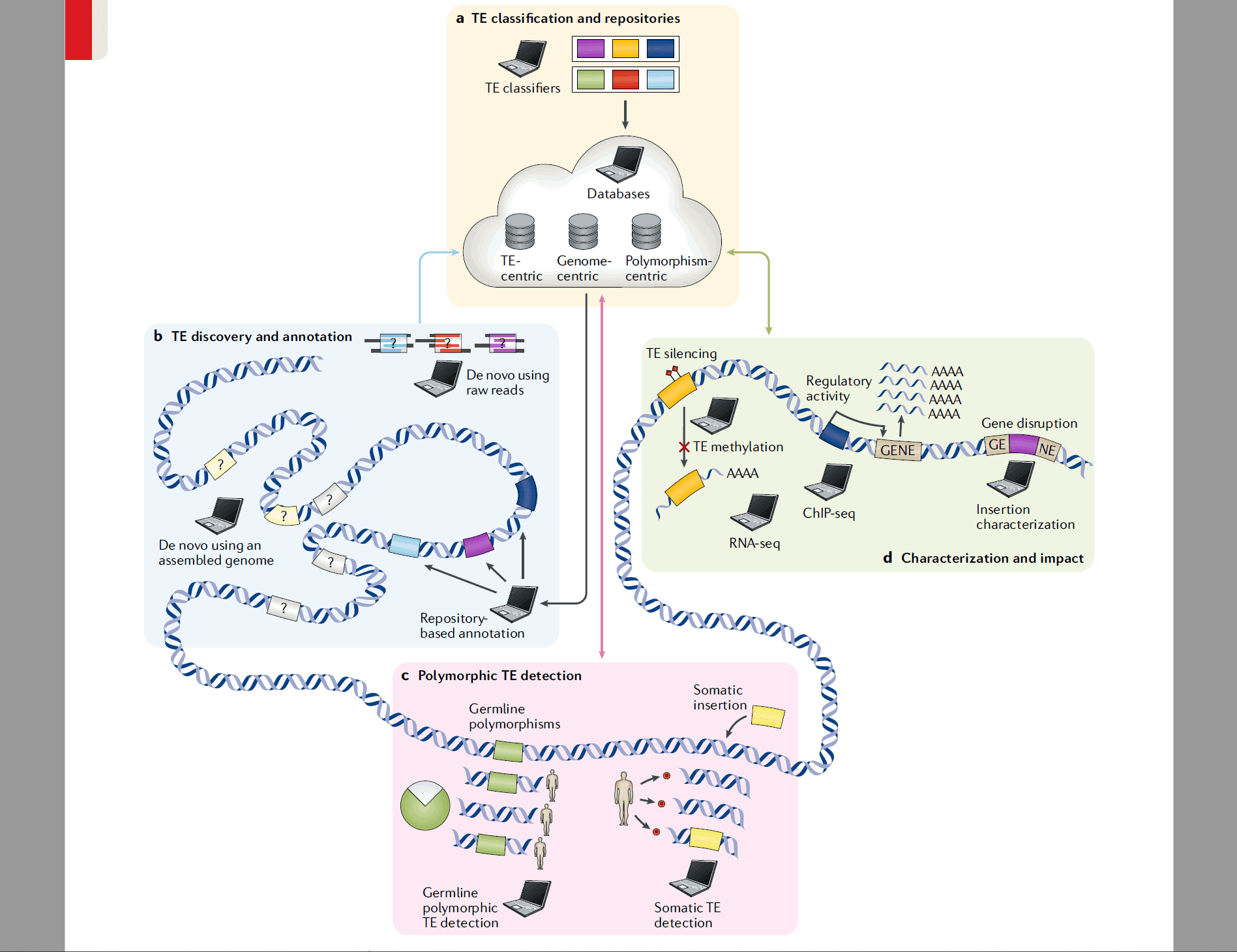
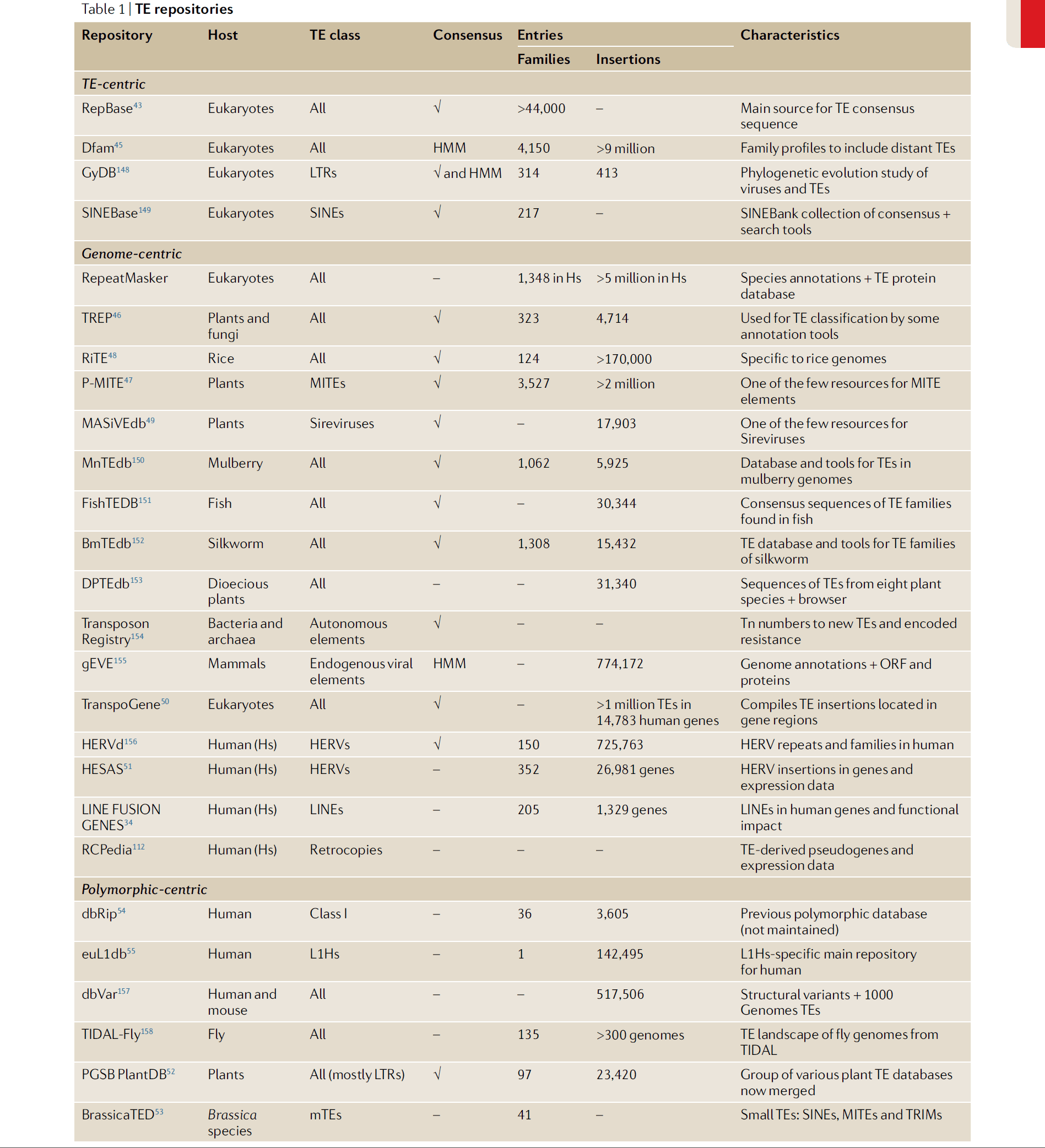
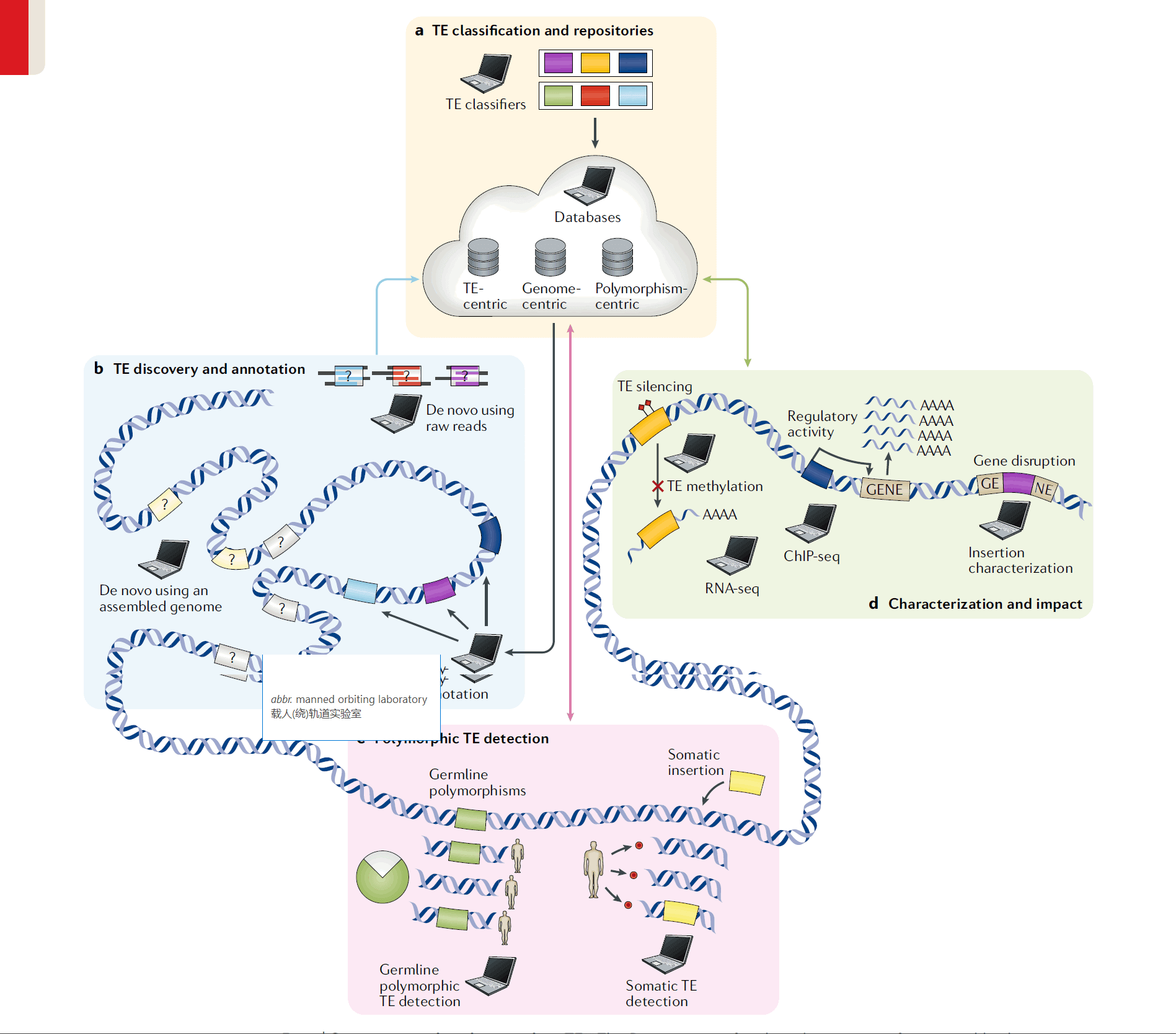
TE分类及数据库
TE分为两大类,有关TE的信息被整合成三类的数据库:
- 以TE为中心的数据库(TE-centric):每个TE家族的保守序列(consensus sequence)
- 以基因组为中心的数据库(genome-centric):特定参考基因组的TE序列
- 以多态性为中心的数据库(polymorphism-centric):基于参考基因组TE多态性插入序列
具体情况如下:
挑战及缺陷
关于TE数据库主要面临两方面的缺陷:
- 物种特异性的TE数据库的建立是很有必要的,数据库之间TE的冗余需要降低以增加内聚性
- 第二个就是人类TE研究中有必要整合TE多态性插入的资源供后续研究
TE构建与注释的手段
转座子序列库的构建方法主要分为三种:
- 从头预测
- 同源预测
- 结构预测
具体的方法以及软件如下:
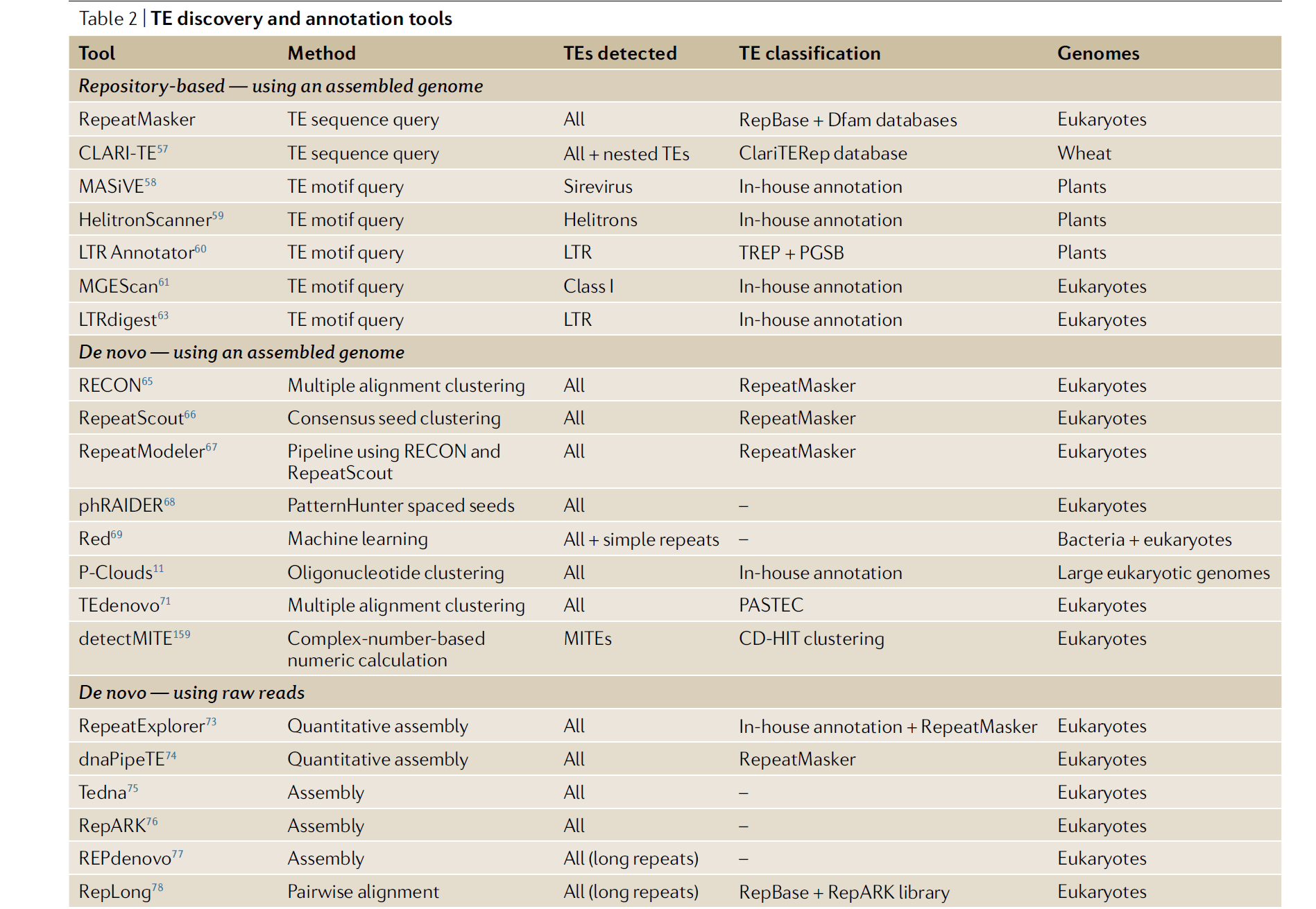
从上表可以看出,实际上就是要么基于数据库要么从头预测。具体注释策略图示如下:
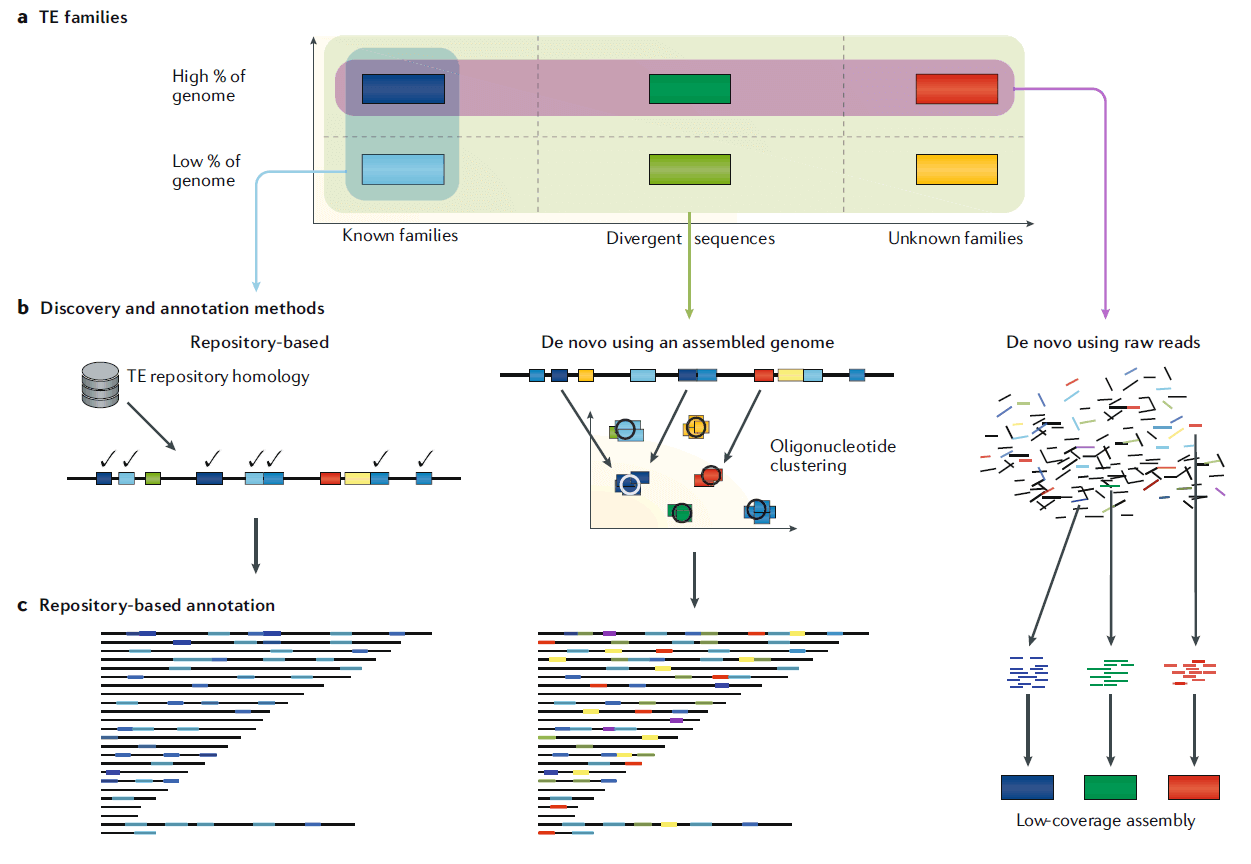
用来分析TEs的计算机工具非常多,如下图所示:
基于数据库注释
这一策略的核心思想是在全基因组范围内对不同TE家族的保守序列以及功能结构域进行检索,RepeatMasker是最常用的工具,是当前TE研究的金标准,基于Repbase以及Dfam数据库。
基于已组装基因组的从头注释
这种方法主要是基于已组装的基因组,可以鉴定一些基于数据库注释缺失的TEs,人类基因组TE通过这种方法从头注释可以鉴定大约多10%的TE。这一方法虽然存在假阳性,但是可以发现新的TE家族以及事件。
基于原始序列的从头注释
这一策略主要充分利用大量的TE序列以及低深度的测序数据去组装TE以及其它重复序列。
挑战以及缺陷
目前TE注释的主流是利用多种策略进行检索并整合到一起,组成比较完整的数据库。比如人类基因组TE数据通过不同策略以及工具注释的结果差异是很大的:
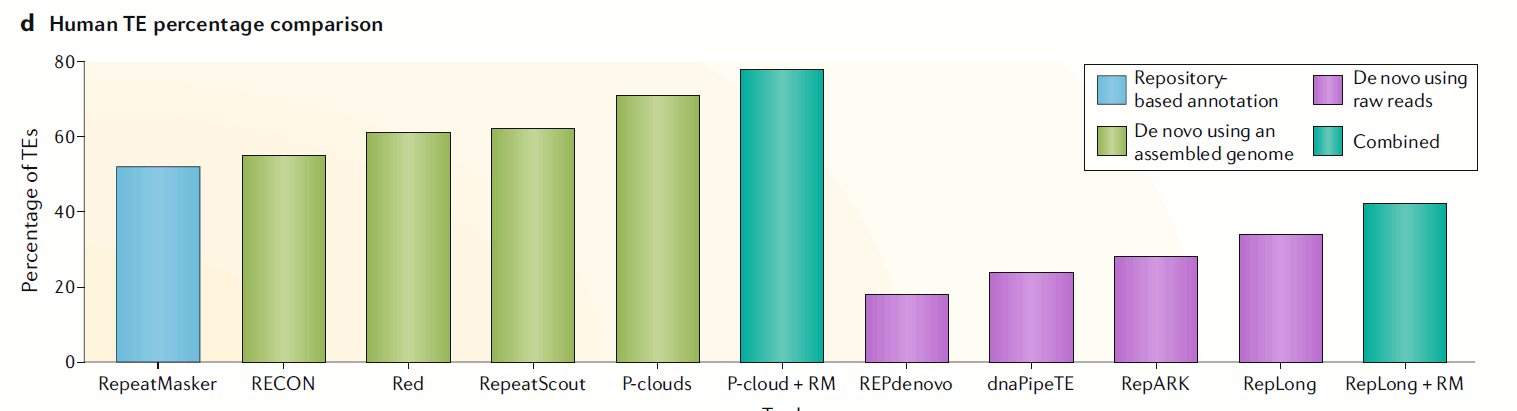
虽然从头注释可以鉴定新的重复序列家族以及新的TE事件,但是引入的假阳性也更高。基于原始序列注释TE的方法只能鉴定少量的TE,但是可以在缺乏组装基因组的情况下注释TE。新的系统性的注释工具对于TE的注释来说极其重要。
TE多态性检测
有了参考基因组、完整的转座子注释文件以及群体就可以进行群体转座子多态性插入的检测,跟SNP一样,TE多态性标记可以跟表型进行关联。目前有大量的软件被开发出来进行TE多态性检测:
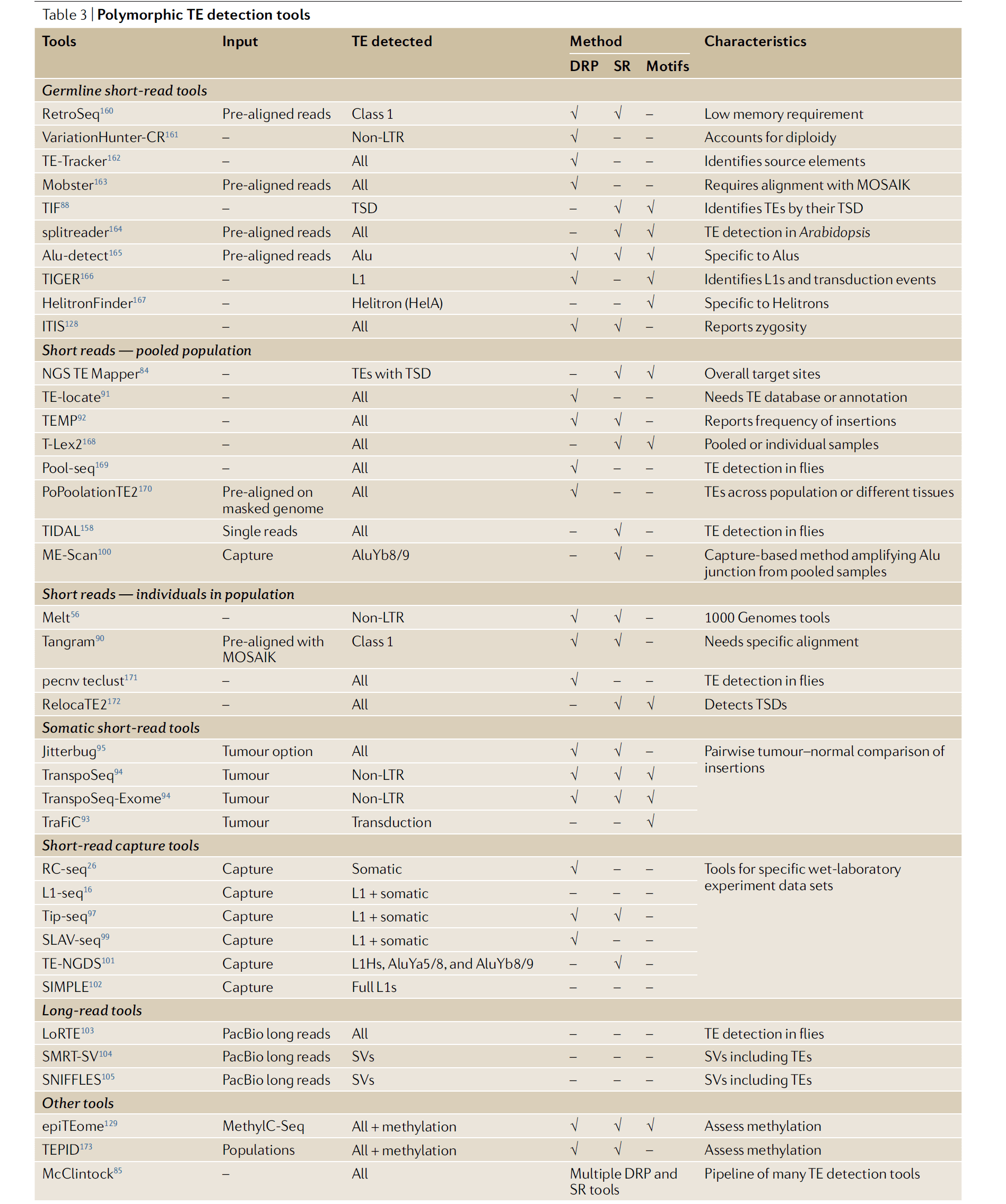
基于短读长序列检测生殖细胞系TE多态性
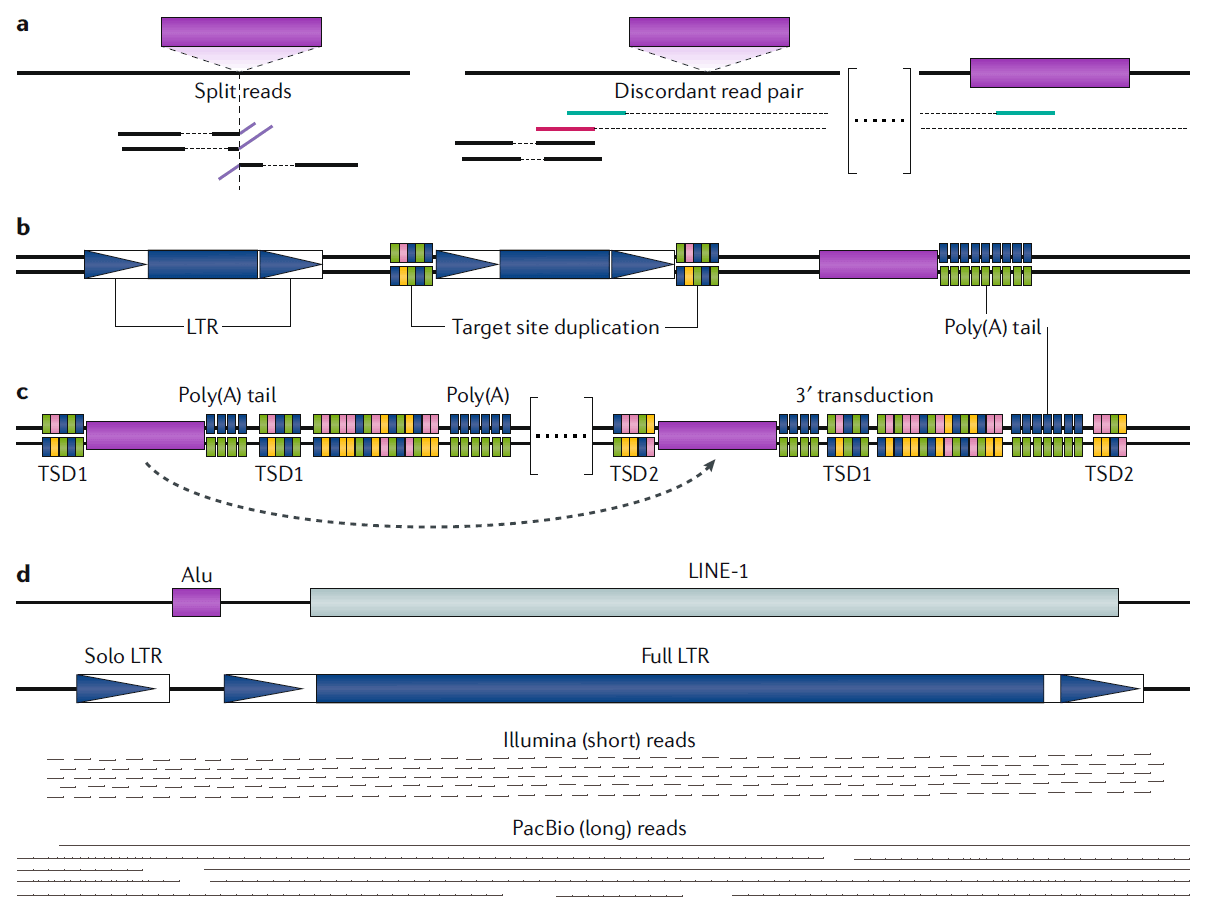
大多数的TE检测流程的输入数据都是NGS测序数据。NGS的读长一般在100-250bp,这对于从属于重复序列的转座子来说,从短读长的reads鉴定TE是一种具有挑战性的工作。目前针对短读长进行TE多态性检测的方法主要有三种:
- 使用split-read (SR)信息
- 依赖不一致的读长对(discordant read pai , DRP)比对
- 基于TE特定motif的识别
TE的特征及影响
TE的插入会引入新的基因或转录本、调控基因表达、导致基因组不稳定以及活跃的置换。
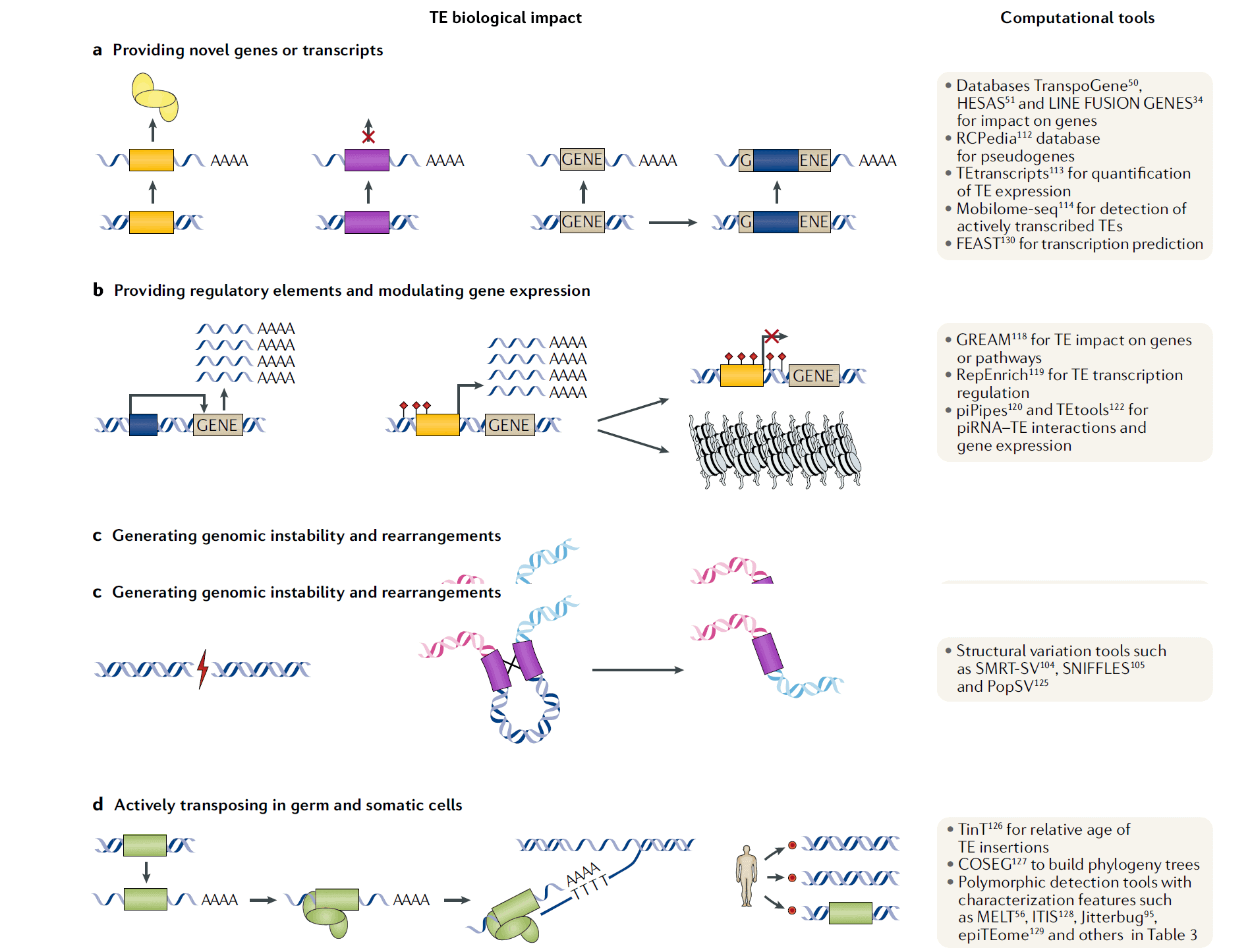
预测TE衍生或者破坏的基因与转录本
注释在基因区域的TE在很多数据库中都有检测到。
预测TE对基因调控的影响
重复区域结构变异的检测
后续还有部分内容暂时这里不讲了,有兴趣的话可以下载 原文细读。