Nature Reviews Genetics | 测序时代的基因组结构变异
简介
结构变异(SV)的鉴定对于解析基因组是十分重要的。得益于检测算法以及测序技术的发展,全基因组结构变异分析成为可能。本文作者综述了基因组结构变异的检测方法,同时指明了整合基因组结构变异以及生物学信息的研究方向。
结构变异
一般是大于50bp的序列变异。SV的大小以及类型多样, 范围从>50bp到~Mb的序列变异。变异类型主要分为:
- 缺失(delections,DELs)
- 插入(insertions,INSs)。根据插入序列可分为移动元件插入(mobile element insertions, MEIs),线粒体基因组插入(nuclear insertions of mitochondrial genome, NUMTs),病毒序列插入(viral element insertions, VEIs)以及其他非特异性插入(insertions of unspecified sequence)
- 重复(duplications, DUPs)。根据重复片段之间的距离可分为串联重复(tandem repeat)以及散在重复(interspersed repeat)
- 倒位(inversions, INVs)
- 易位(translocations,TRAs)
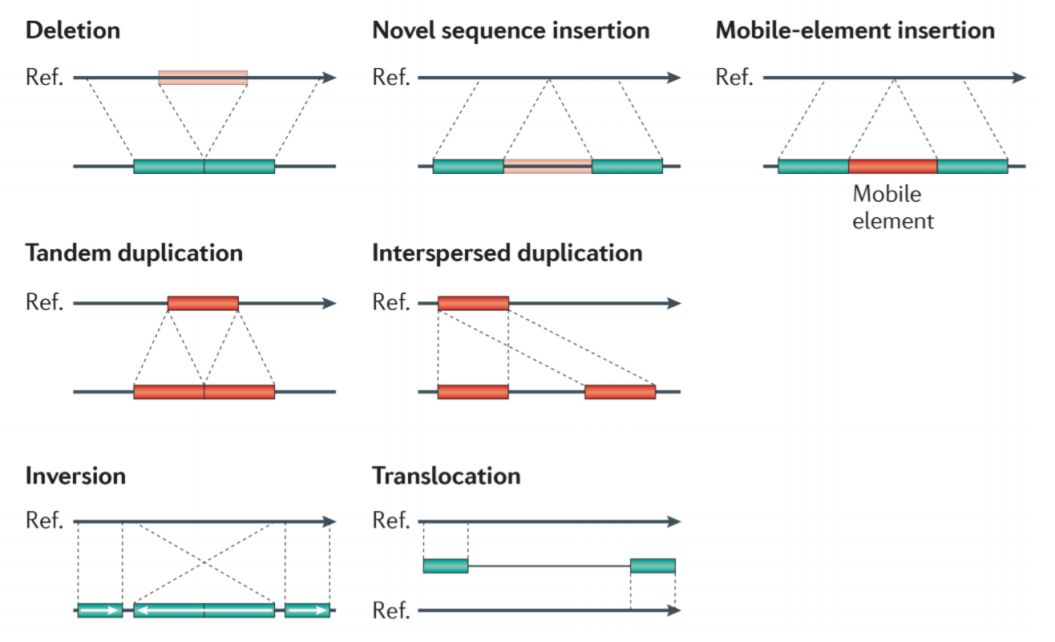
拷贝数变异(copy number variation, CNV)是DELs以及INSs的合称。INVs与TRAs不会改变基因组大小,其他的会改变基因组大小。
SV分布在整个基因组中并影响分子及细胞进程,调节功能,3D基因组结构以及转录机制等。
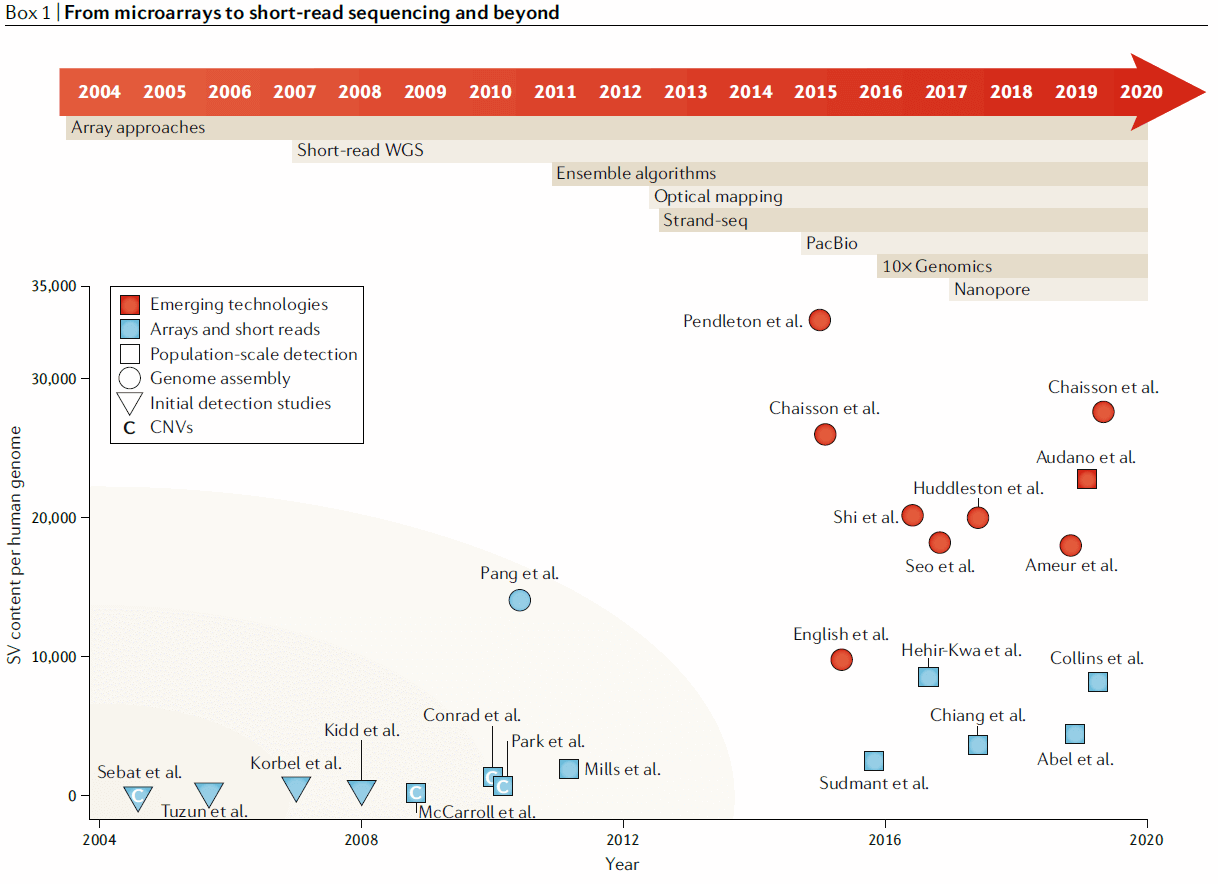
随着检测算法的开发以及测序技术的发展,越来越多以前未被检测到的SV被检测到,从微阵列到短读长测序再到现在的三代长读长测序以及未来新的测序技术,SV的研究如下图所示:
在这篇综述中,作者讨论了解决人类基因组中SV的方法,这些方法通过算发集成以及新技术来绕过短读长的局限性。
算法集成
基于序列的SV检测主要是利用样本序列与参考基因组序列之间mapping不一致产生的位置特征。目前基于NGS数据检测SV的方法主要有四类:
- Read Pair,一般称为Pair-End Mapping,简称RP或者PEM
- Split Read,简称SR
- Read Depth,简称RD,也有人将其称为RC——Read Count的意思,它与Read Depth是同一回事,顾名思义都是利用read覆盖情况来检测变异的方法
- 序列从头组装(de novo Assembly, 简称AS)的方法。
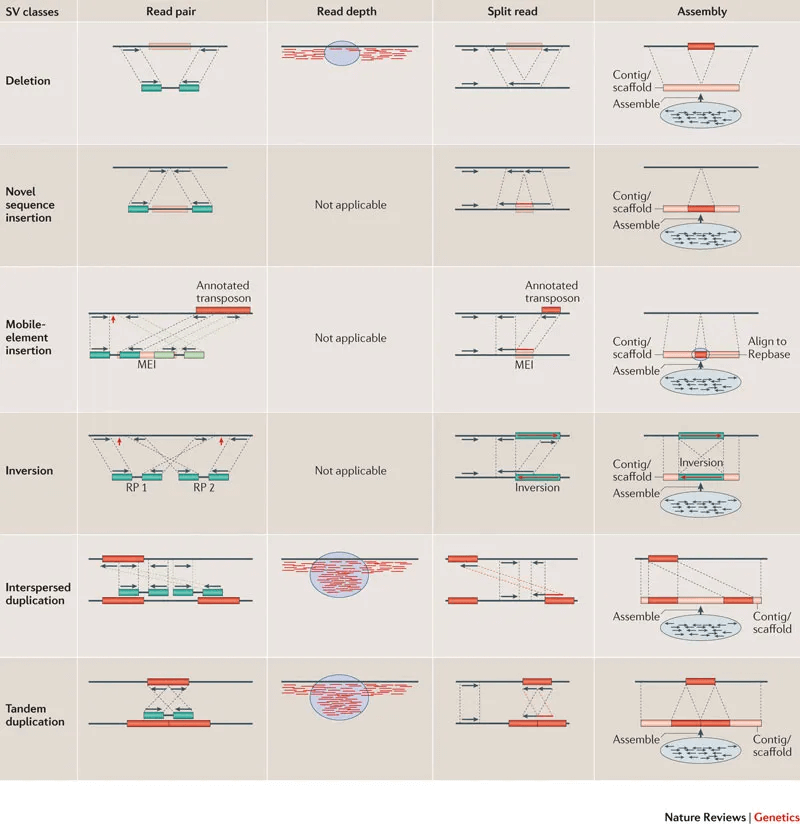
目前大部分的SV检测算法都是涉及到上述一至两类,这样就会导致检测到的SV数目以及准确度都会大打折扣。一种解决办法就是同时利用多种算法也就是上面我们提到的集成算法检测SV再整合到一起形成一个统一的变异数据集,具体操作如下:
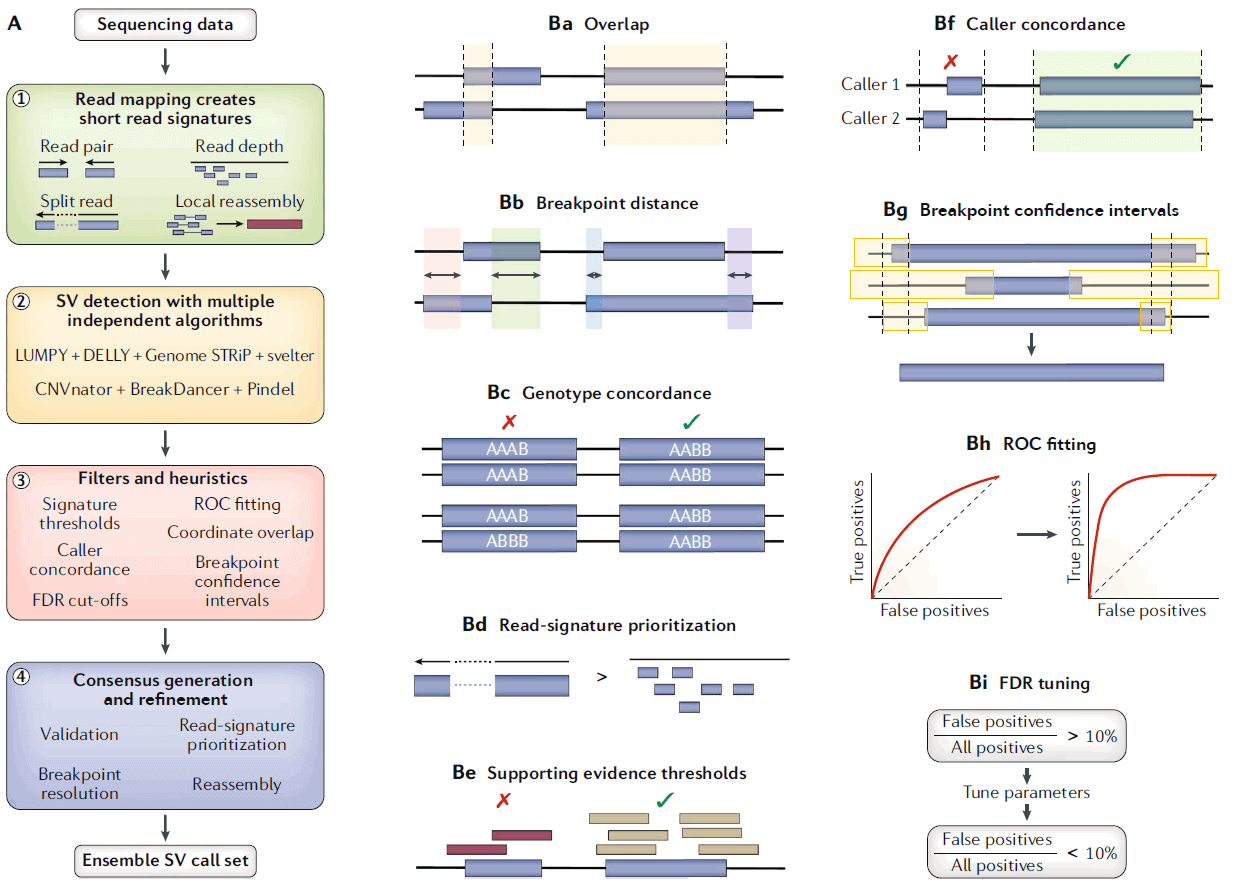
大多数的检测算法都是不同的实验室内部针对某个项目进行开发的,因此就缺乏广适性。但是同时也存在几种算法主要针对上述提到的四类中的一类,这就使得我们可以组合起来就行SV的检测。比如CNVnator可以与BreakDancer和Pindel组合分别针对cover read depth, read pair and split reads进行SV的检测。同时为了降低假阳性还需要设置一系列的过滤指标。
大规模群体SV检测
大规模群体检测SV的研究目前有很多,比如我们熟知的1000 Genomes Project (1KGP)整合了19中算法进行SV的检测。
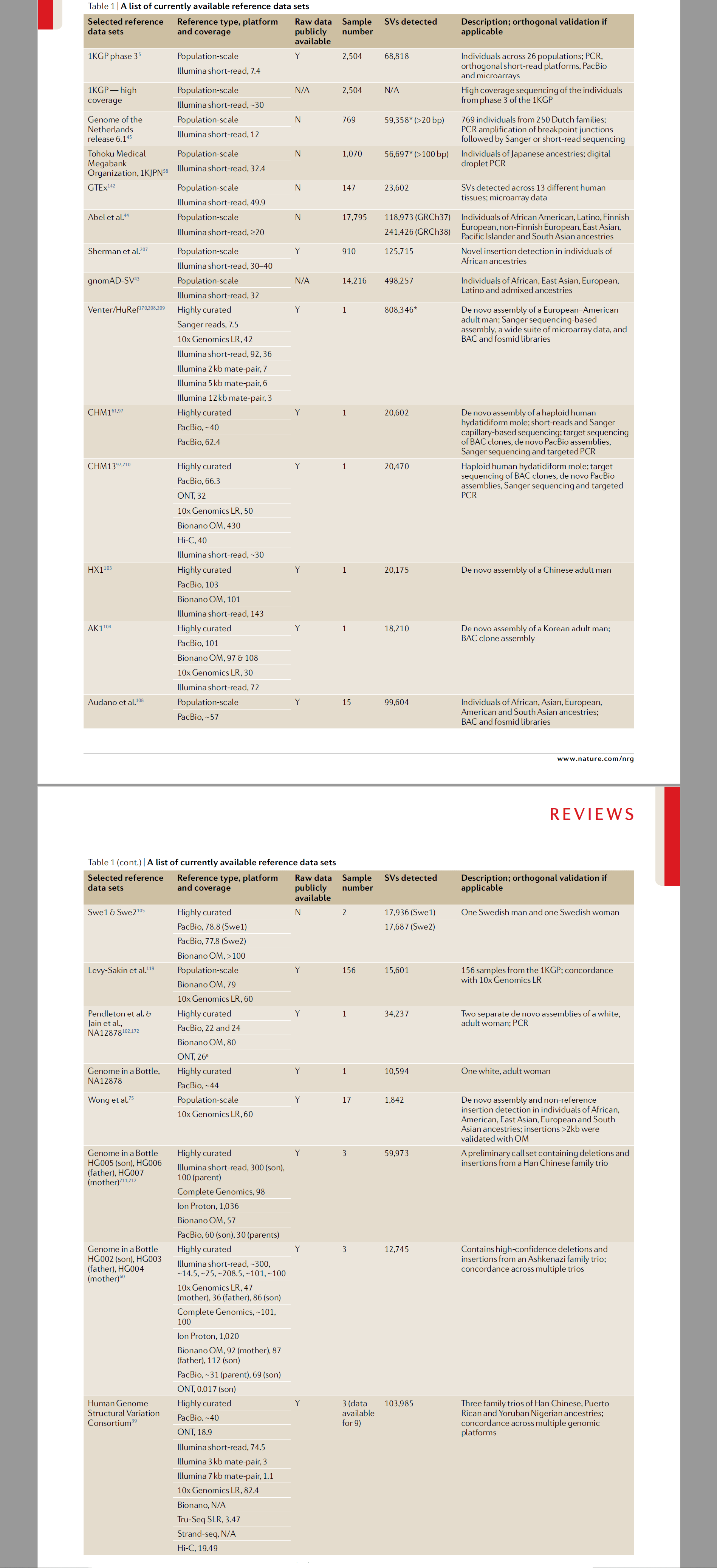
新兴基因组技术
大量的新兴技术试图超越二代测序短读长的限制。连接分子策略比如链读长、链式测序以及Hi-C等技术通过推断远端定位的序列比对进行SV的检测。单分子测序目前主要是Pacbio以及Nanopore。具体如下:
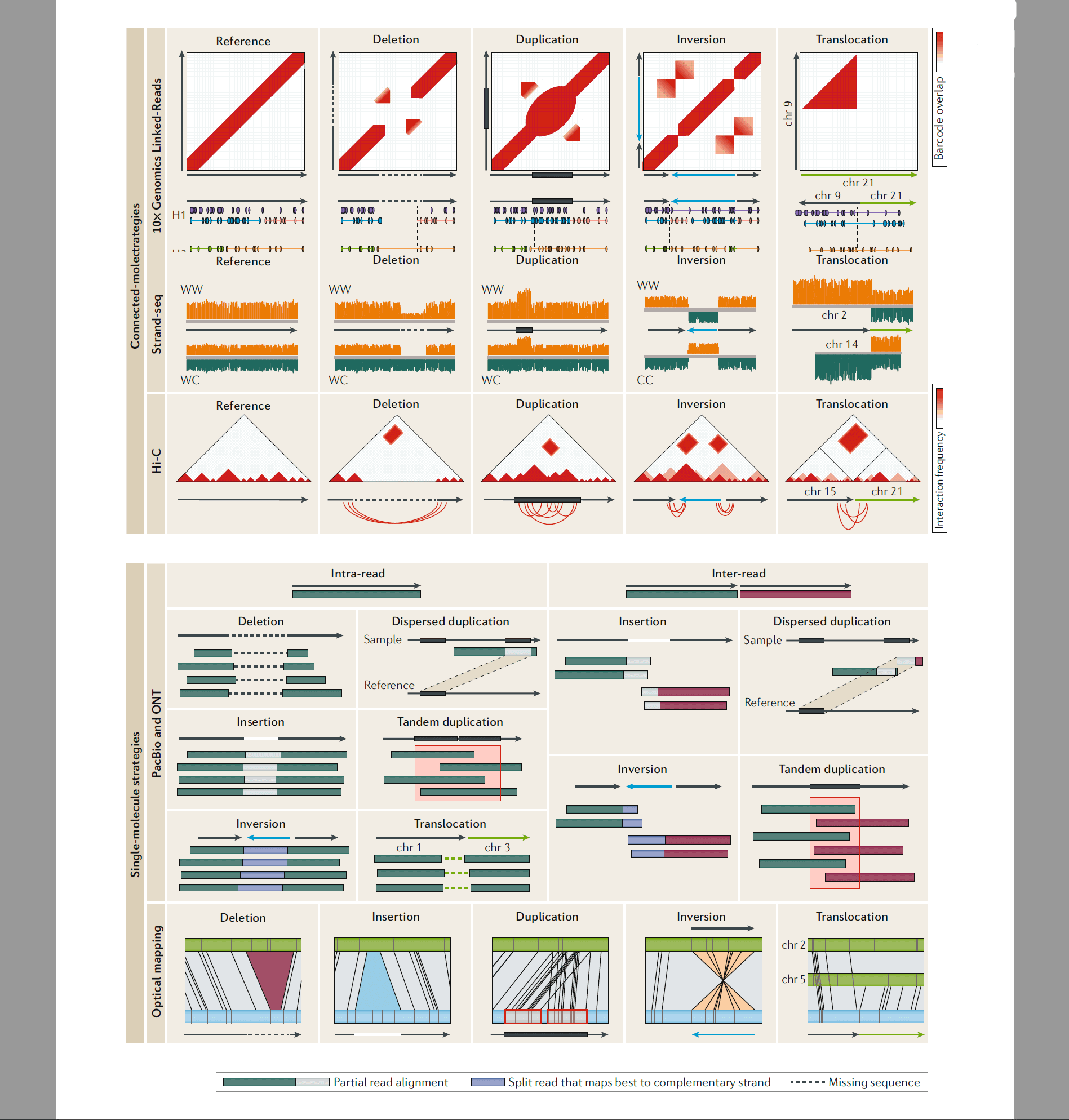
具体的这里不展开,有兴趣的可以下载 原文仔细阅读。
总结来说,目前检测SV的方法及软件汇总如下,最好的策略肯定是利用单分子三代测序比如Pacbio或者Nanopore,但是价格感人,所以前期积累的大量的二代高深度测序数据就可以利用起来了,针对二代测序推荐使用SppedSeq,整合多种算法,覆盖了多种SV类型的检测。
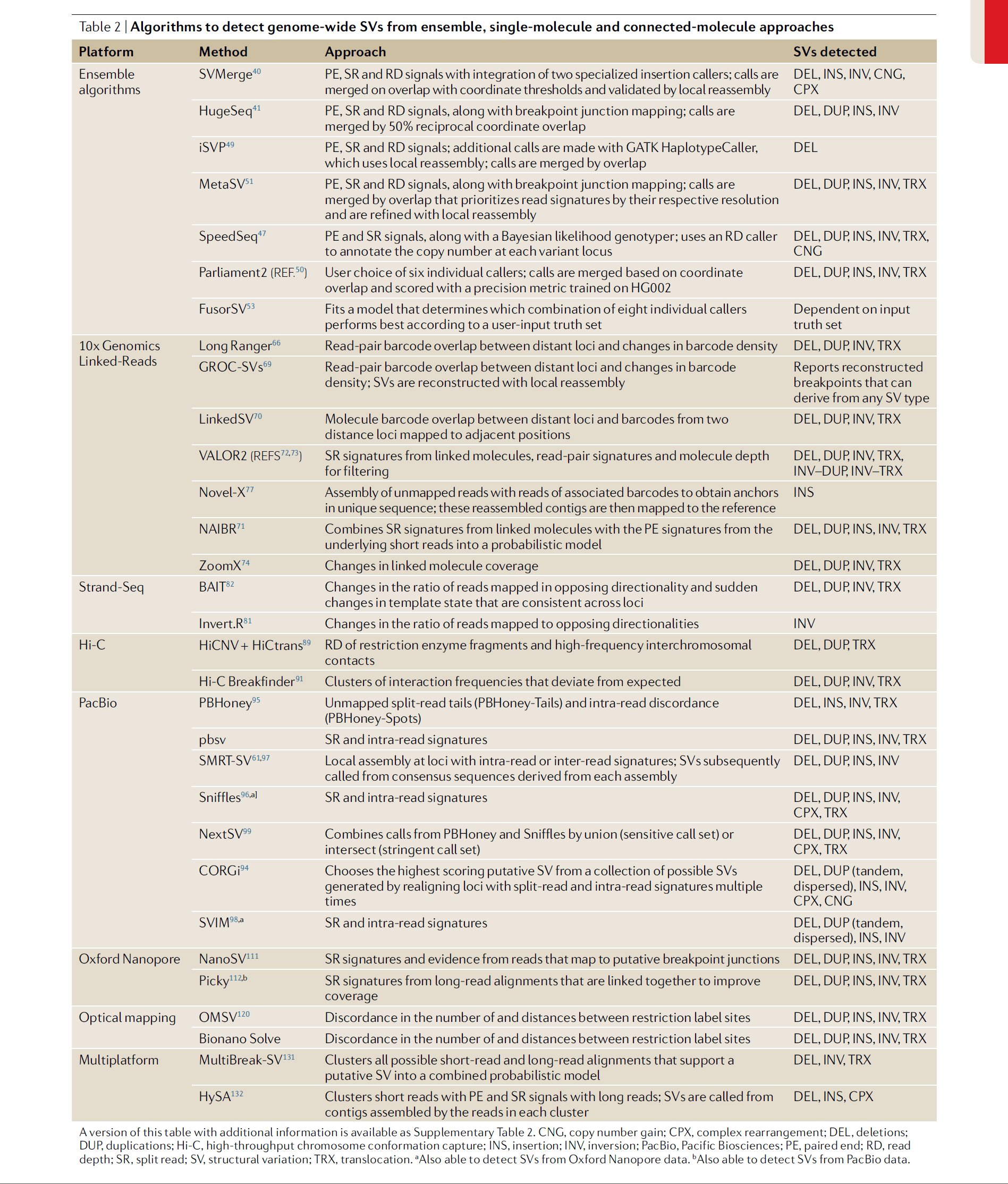
这篇综述内容非常广,涉及面也多,有兴趣的话可以仔细阅读,我这里只是粗略读了一下,主要是为了找几种比较好使的SV检测软件。